血友病是一种遗传性凝血功能障碍的疾病,主要是凝血酶生成障碍,凝血时间长,终身具有轻微创伤后出血倾向,重症患者没有明显外伤也会自发性出血。
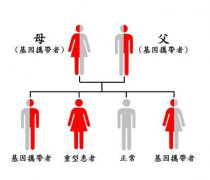
血友病是一组先天性凝血因子缺乏,致出血性的疾病,先天性因子Ⅷ缺乏为典型的性联隐性遗传,由女性传递,男性发病,控制因子Ⅷ凝血成分合成的基因位于X染色体,患病男性与正常女性婚配,子女中男性均正常,女性为传递者;正常男性与传递者女性婚配,子女中男性半数为患者,女性半数为传递者;患者男性与传递者女性婚配,所生男孩半数有血友病,所生女孩半数为血友病,半数为传递者,约30%无家族史,其发病可能因基因突变所致。
常见的血友病类型:
1.血友病A(血友病甲),即因子Ⅷ促凝成分(Ⅷ:C)缺乏症,也称AGH缺乏症,是一种性联隐性遗传疾病,女性传递,男性发病。
2.血友病B(血友病乙),即因子Ⅸ缺乏症,又称PTC缺乏症、凝血活酶成分缺乏症,亦为性联隐性遗传,其发病数量较血友病A少。但本型中有出血症状的女性传递者比血友病A多见。
3.血友病C(血友病丙),即IXa因子缺乏症,又称PTA缺乏症、凝血活酶前质缺乏症。为常染色体不完全隐性遗传,男女均可患病,是一种罕见的血友病。
15~20/10万男孩中有发病,此发病率在所调查的不同的种族和地域之间没有差异。发病率以血友病A最多占85%,血友病B占15%,血友病C较少见。

那么血友病A都有那些临床表现和并发症呢?
1.出血
为本病主要的表现。终身有轻微损伤或手术后长时间出血的倾向。出血程度及发病的早晚与患者血浆中FⅧ活性水平有关。根据出血轻重与血浆中凝血因子活性的水平,将本病分为4型:
(1)重型 血浆中FⅧ活性<1%,常在2岁以前就出血,在婴儿开始学爬、学走后出现出血症状,甚至结扎脐带时出血不止。患者出血部位多且严重,常有皮下、肌肉及关节等部位的反复出血,关节内血肿畸形多见。此外,还可见肾脏出血导致血尿、胃肠道出血、腹腔内出血,肺、胸腔、颅内出血少见。
(2)中间型 FⅧ活性为1%~5%,起病在童年时期以后,以皮下及肌肉出血居多,亦有关节出血,但反复次数较少,严重程度也轻于重型。
(3)轻型 FⅧ活性为5%~25%,出血多在青年期,由于运动、拔牙或外科手术后出血不止而被发现,出血轻微,可以正常生活,参加运动,偶尔发生关节血肿。
(4)亚临床型 只有大手术后才发生出血,实验室检查可以证实为本病,FⅧ活性为25%~40%。
一般而言,凡出血症状出现越早,病情越重,随年龄的增长,出血症状可逐渐减轻,有时可出现无出血症状的缓解期。出血可在创伤后数小时或数天后发生,也可在创伤或手术后即渗血不止。
2.出血所导致的压迫症及并发症
出血形成血肿后可导致压迫症状:
(1)周围神经受累 发生率为5%~15%,病人有麻木、剧痛、肌肉萎缩。
(2)上呼吸道梗阻 口腔底部、喉、舌、扁桃体、后咽壁或颈部的严重出血甚为危险可引起窒息。
(3)压迫附近血管 可发生组织坏死。
那么有血友病遗传基因的家庭就不能生出健康的宝宝了吗?
齐康喜孕多专家团告诉您:可以!血友病是属于单基因遗传病,是由单个致病基因所导致的遗传病,完全可以通过第三代试管婴儿PGD技术生出健康的宝宝。PGD是胚胎种植前基因诊断,主要用于检查胚胎是否携带有遗传缺陷的基因。在精子卵子结合形成受精卵并发育成胚胎后,在其植入子宫前使用PGD技术进行基因检测,以便使体外授精的试管婴儿避免一些遗传疾病。目前植入前遗传诊断能诊断一些单基因缺陷引发的疾病,比如说地中海贫血症血友病等疾病,从而生出健康的宝宝。





{kind=link}